Note: If you’re trying to follow my work chronologically, this is really more like WGCNA 5 - I did a small recap of some WGCNA work in my December Goals post (the previous one).
Regardless, this post summarizes a few changes I’ve made to my WGCNA analysis recently. They are as follows:
- Changing Hematodinium qPCR levels
- Adding new analyses to compare change in Hematodinium qPCR levels
- Removing irrelevant analyses
Changing Hematodinium qPCR levels
Process Description
As part of my WGCNA analysis (again, using signed networks), I wanted to examine Hematodinium levels within the crab. To get a number for this, I took the SQ mean from all runs for that crab. However, qPCR was ran on samples from Day 0 and Day 17, and often the Hematodinium level changed substantially between those two timepoints. Rather than averaging the SQ mean between Days 0 and 17 (if available for that crab), I decided it would be optimal to use Day 0 SQ mean only as my WGCNA input. This had two benefits:
-
1: Removes issue of elevated-temperature crab, for which only Day 0 qPCR data is available
-
2: Removes conflation of Hematodinium infection direction. You’d expect a sample with rapidly increasing Hematodinium to have markedly different expression from a sample with markedly decreasing Hematodinium, but it’s quite possible that this would be covered up by taking the mean.
To preserve previous runs (just in case I wanted to look at them again later), I put them in a subdirectory called “hemat_average”)
Prior to re-running WGCNA, I went back and looked at Pam’s qbit results. I’m glad I did, because I found an error in my sheet! Not sure where it originated (maybe simple copy-paste error or typo, maybe something in the project sheet I got from grace) despite poking around and trying to find the error in the original, but regardless, for Crab E, the SQ Mean values for Day 0 and Day 17 were reversed! Luckily not an issue for any analyses previously, but definitely good to catch because of the new analyses I’ll describe in the next section.
Results
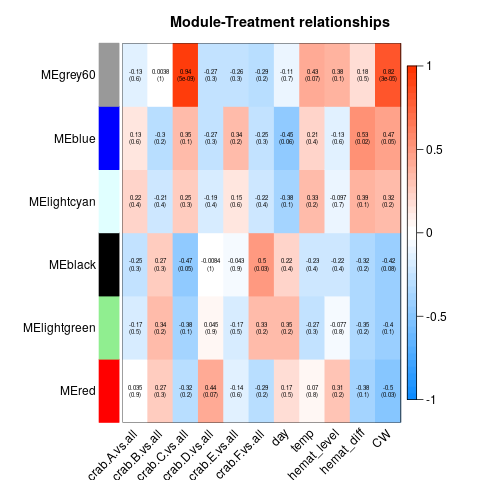
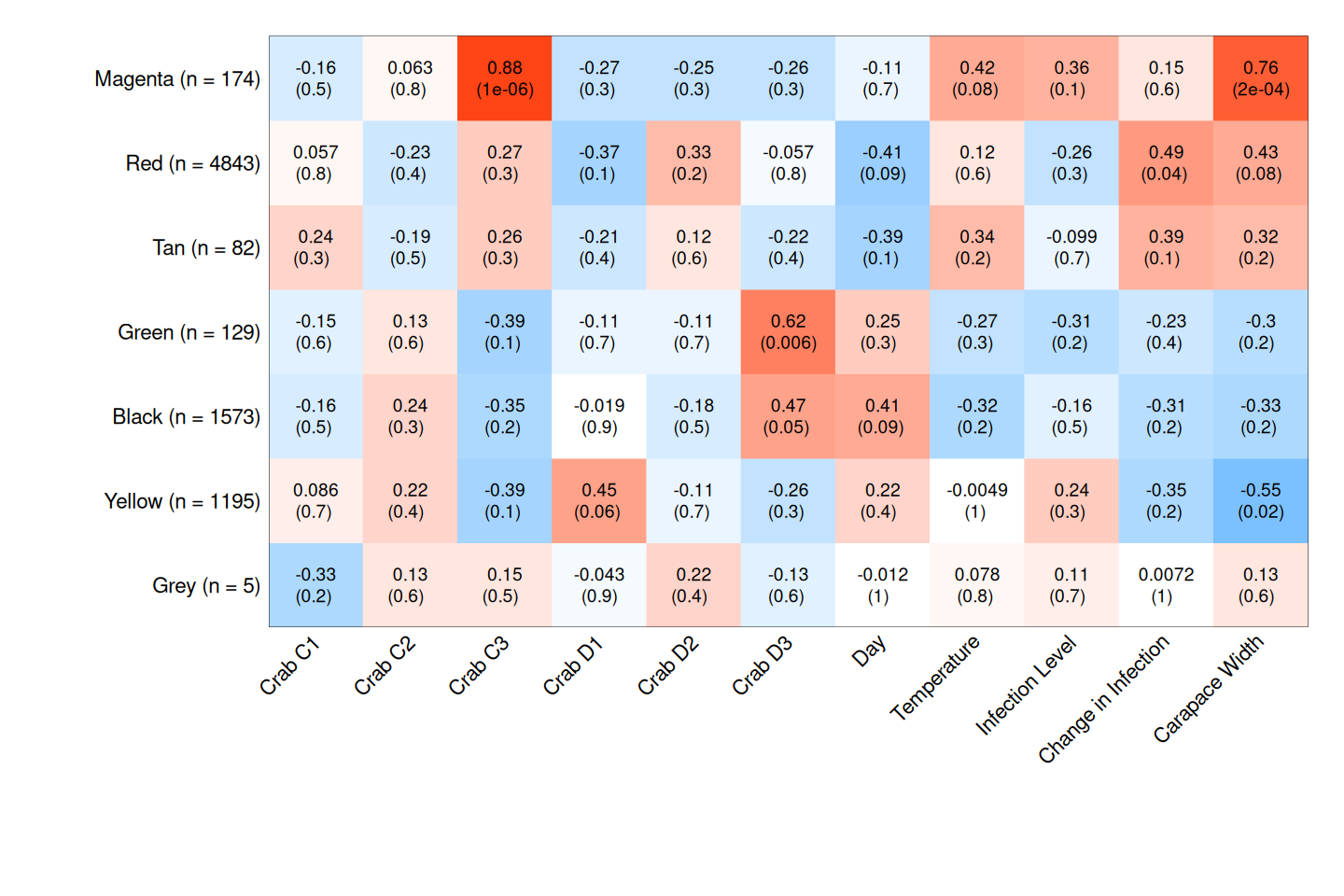
Again, I ran WGCNA on samples from all crabs on all days. Variables were crab, day, temperature (with pairwise comparisons and vs. all), Hematodinium level on Day 0, carapace width, and shell condition. Here’s the heatmaps from the complete, Tanner crab, and parasite transcriptomes, respectively:
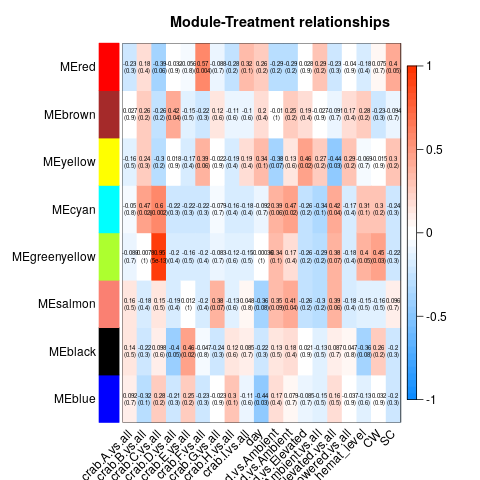
As before, I examined module/variable correlation when correlation was <= 0.05. If the correlation appeared to be predominantly due to the effect of a single crab, I discarded it. That left me with the following modules and correlations:
| Transcriptome | Module | Trait(s) | p-value(s) |
|---|---|---|---|
| Complete | yellow | Lowered vs. Elevated, Elevated vs. All | 0.02, 0.03 |
| Complete | cyan | Lowered vs. Ambient, Elevated vs. All | 0.02, 0.04 |
| Complete | salmon | Lowered vs. Ambient | 0.04 |
| Complete | blue | day | 0.03 |
| Tanner crab | black | Lowered vs. Ambient, Elevated vs. All | 8x10^-4, 0.05 |
| Tanner crab | brown | Lowered vs. Elevated, Elevated vs. All | 0.02, 0.03 |
| Parasite | brown | Hematodinium level | 0.02 |
| Parasite | pink | day | 0.03 |
My next task is to run these back through GO-MWU to see if anything interesting pops out!
New analysis to compare change in Hematodinium levels
Previously, I treated infection level as a constant. However, we do have two timepoints here - the start and end of the experiment, Days 0 and 17. Therefore, it makes a whole lot of sense to examine change in infection level as a variable! There is a complicating factor. Since all elevated-treatment crabs died before Day 17 of the experiment, we only have qPCR data from a single timepoint. Therefore, I restricted this analysis to Lowered and Ambient-temp crabs only.
Essentially, to calculate change in Hematodinium level, I took the SQ Mean from Day 17, divided it by the SQ Mean from Day 0 for that same crab, and then put it on a log scale. I then added those numbers in as the variable hemat_diff, and ran WGCNA again!
This run of WGCNA included slightly different variables.
- Added hemat_diff, as described above
- Removed hemat_level, since I wanted to examine the change rather than the starting point
- Removed Shell Condition as a variable, since only one crab had a shell condition other than New (and I’m already looking at the effect of each crab individually)
- Dropped pairwise comparisons and vs. all for temperature comparison, since dropping the Elevated group changes it to a pairwise comparison already
Otherwise, pretty much everything about my run setup was the same!
Results
Naturally, removing Elevated-temperature samples decreased my sample size. Therefore, I ignored correlation between modules and variables other than hemat_diff. After all, I had already looked at them (and more accurately) in my previous run on all samples!
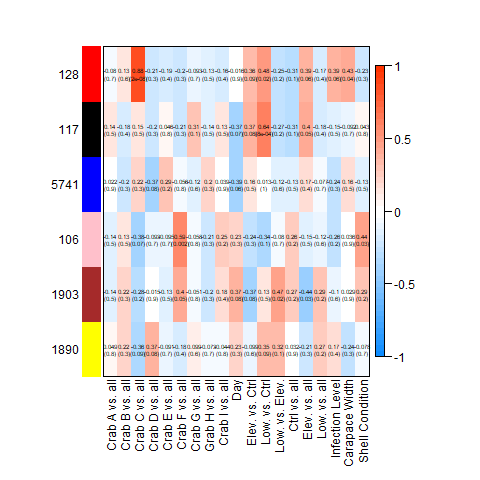
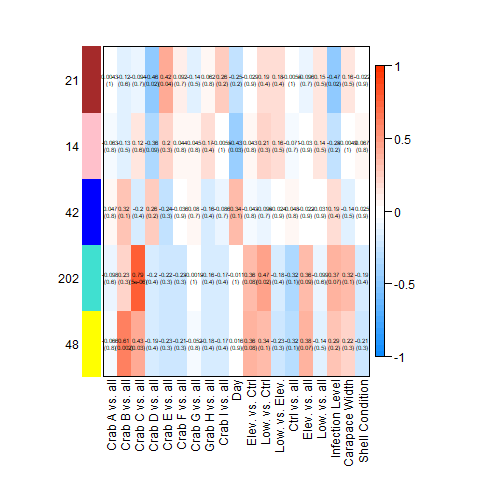
Still, I’ll post the heatmaps from the complete, Tanner crab, and parasite transcriptomes, respectively:
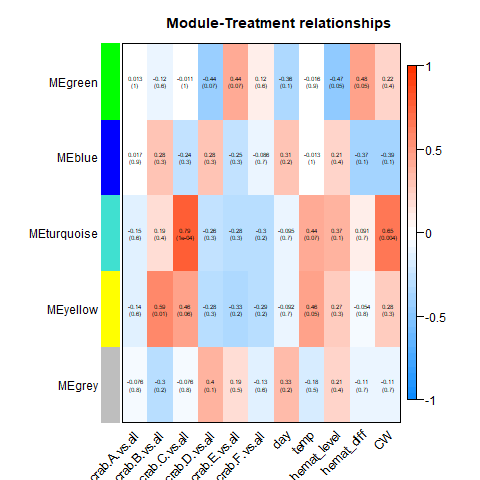
After discarding modules linked strongly to individual crab, as described above, this left me with the following modules associated with hemat_diff:
| Transcriptome | Module | p-value |
|---|---|---|
| Complete | blue | 0.02 |
| Tanner crab | red | 0.04 |
| Parasite | green | 0.05 |
For all, trait is hemat_diff (I ignored any correlation with other traits for reasons described above).
Next step is to run these guys through GO-MWU to see if anything interesting comes out!
Removing irrelevant analyses
As I was going through my scripts and output folders, I noticed I had a whole bunch of old WGCNA runs. Most of them were outdated or irreparably broken for some reason or another. For instance, I did the following:
-
Analyzed only elevated- or ambient-temp crab (sample size of 6-9) when the minimum sample size for WGCNA is ~15
-
Tried to perform pairwise comparisons with separate WGCNA runs instead of putting in all samples and letting WGCNA make the pairwise comparisons
-
Set hemat_level (SQ mean from qPCR) as high/low rather than using the precise number
-
Used an unsigned instead of signed network - signed is typically optimal as described here
So yep, just a whole mess of mistakes! I removed irrelevant scripts and their outputs, and renumbered the existing scripts (remember, all of which begin with 5_) to fix this.